QMSR Implementation & Inspection Readiness: A Comprehensive Fact-Check and Operational Analysis of the February 2, 2026, Transition
The transition to QMSR is not merely a bureaucratic exercise in updating citations; it represents a fundamental philosophical pivot from a compliance-based model to a risk-centric, Total Product Life Cycle (TPLC) regulatory framework.4
Executive Summary: The "Day One" Reality of Harmonization
As of today, February 2, 2026, the regulatory landscape for medical device manufacturers in the United States has undergone its most profound structural shift in three decades.1 The two-year transition period following the publication of the Quality Management System Regulation (QMSR) final rule has expired. The Food and Drug Administration (FDA) has officially begun enforcing the amended 21 CFR Part 820, effectively retiring the Quality System Regulation (QSR) that had governed the industry since 1996.1 This date also marks the definitive withdrawal of the Quality System Inspection Technique (QSIT), the inspection methodology that shaped the careers of a generation of quality professionals, replacing it with the Inspection of Medical Device Manufacturers Compliance Program (CP) 7382.850.1
This report serves as an exhaustive operational guide and a rigorous fact-check of drafted advisory content for qms.coach. It is designed to validate strategic advice against the hard reality of the final rule and the newly implemented inspection protocols. The transition to QMSR is not merely a bureaucratic exercise in updating citations; it represents a fundamental philosophical pivot from a compliance-based model to a risk-centric, Total Product Life Cycle (TPLC) regulatory framework.4
The immediate and most critical operational change is the removal of the 21 CFR 820.180(c) exemption.6 For the first time, FDA investigators possess the unequivocal legal authority to inspect management review minutes, internal audit reports, and supplier audit records—documents previously shielded to encourage internal candor.6 This regulatory transparency creates unprecedented vulnerability for manufacturers who have relied on the "glass wall" between internal quality discussions and external regulatory scrutiny.
Furthermore, this report contextualizes the QMSR enforcement within the broader "regulatory storm" of 2026. As US manufacturers navigate "Day One" of QMSR, they are simultaneously racing toward the May 28, 2026 deadline for mandatory EUDAMED module usage in the European Union.8 The convergence of these timelines signifies the end of localized compliance strategies and the beginning of a harmonized, transparent global quality era.

Part I: Fact-Checking the qms.coach Strategy
A primary objective of this report is to validate the insights and warnings drafted for the qms.coach blog series. The drafted content addresses critical transition themes, ranging from supplier quality to inspection readiness.9 Our analysis confirms that while the blog's strategic direction is sound, the nuances of the final rule and the newly released inspection guidance (CP 7382.850) require a deeper level of technical precision to ensure full compliance.
1.1 Topic Validation: "Building Your First QMSR-Compliant QMS from Scratch"
The blog posits that startups possess a strategic advantage by building a QMSR-native system rather than retrofitting a legacy QSR system.10
- Verdict: Accurate and Critical.
- Analysis: Legacy quality systems built on the 1996 QSR are often heavily siloed, with distinct procedures for "Design Control" (820.30) and "Production" (820.70) that may not naturally integrate the "Risk-Based Approach" required by ISO 13485:2016. Startups have the luxury of adopting the process-based approach of ISO 13485 from inception, where risk management (ISO 14971) is woven into the fabric of every SOP rather than treated as a standalone deliverable.11
- Nuance Added: The "Start from Scratch" advantage is only realized if the startup simultaneously addresses the Subpart B additions (21 CFR 820.35). A purely "off-the-shelf" ISO 13485 system will fail if it does not explicitly incorporate the US-specific requirements for complaint files, servicing records, and UDI tracking.12 The "advantage" is in the structure, but the content must still be a hybrid model.
1.2 Topic Validation: "QMSR Compliance: Common Mistakes"
The blog identifies "treating QMSR as a documentation exercise" and "failure to separate CAPA" as primary pitfalls.14
- Verdict: Accurate and High Risk.
- Analysis: The shift from QSR to QMSR is frequently misunderstood as a simple "Find and Replace" exercise (e.g., swapping "DHF" for "Design and Development File"). This superficial compliance ignores the cultural shift demanded by the FDA's new "Culture of Quality" inspection objective.5
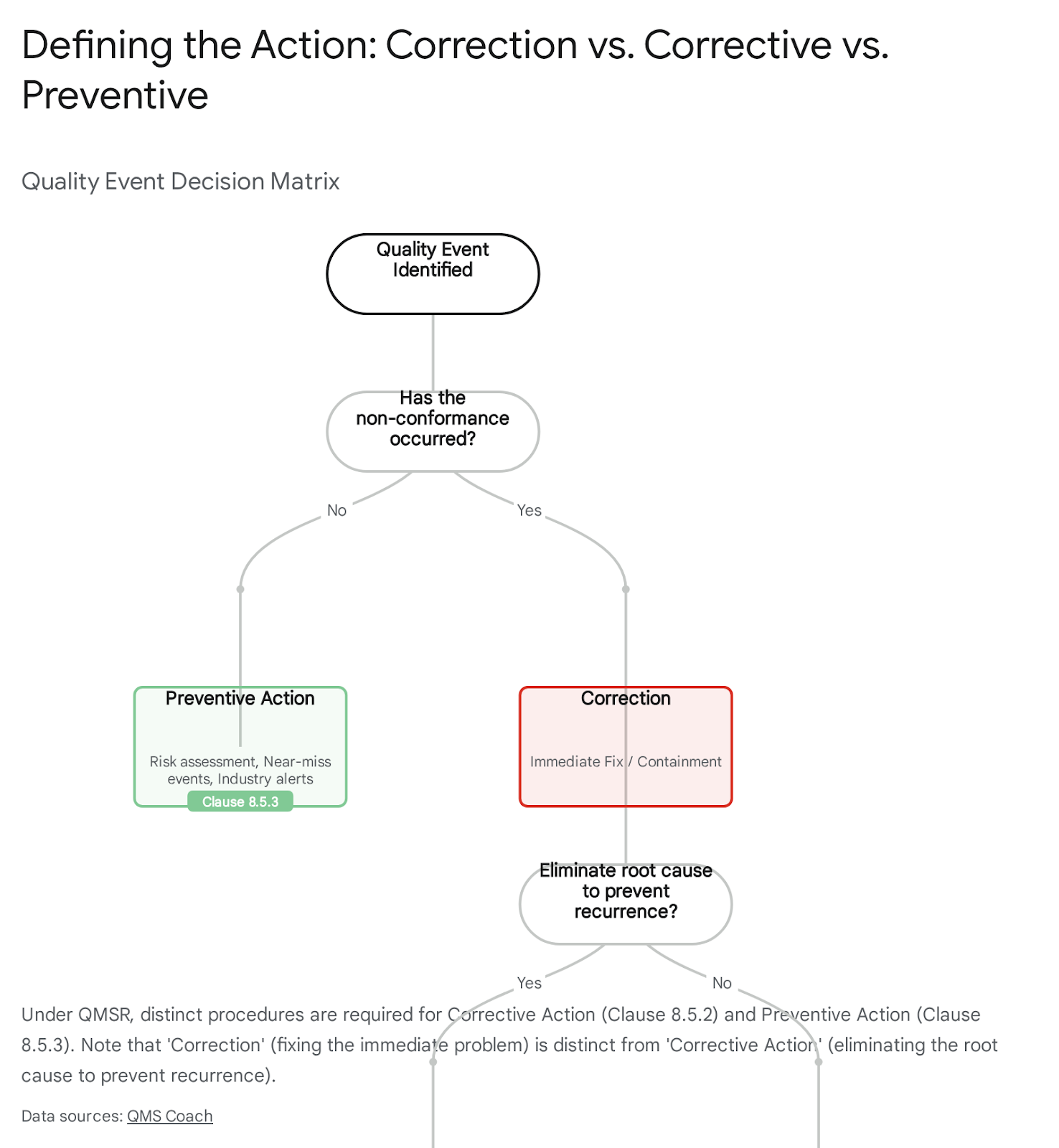
- CAPA Nuance: The blog correctly identifies the danger of commingling Corrective and Preventive Actions. Under the old QSR 820.100, these were often treated as a single workflow. ISO 13485 (and thus QMSR) separates them into Clause 8.5.2 (Corrective) and Clause 8.5.3 (Preventive).14 The FDA's new inspection program will look for distinct data analysis triggers for each. A system that processes "potential" problems through a "reactive" corrective action pathway will distort the data analysis required by Clause 8.4, leading to findings on the effective use of quality data.
1.3 Topic Validation: "What to Expect in Your First QMSR Inspection"
The blog warns that inspections will look different, with inspectors referencing ISO clauses and requesting previously exempt records.9
- Verdict: Accurate but Understated.
- Analysis: The change is more than just referencing clauses; it is a total restructuring of the inspection flow. The withdrawal of QSIT means the "four subsystem" model is gone. It is replaced by a "six area" model that is circular and risk-driven.4 The blog correctly identifies the risk of record transparency (internal audits), but the implication is profound: inspectors will likely use internal audit findings as a roadmap for their own investigation. If an internal audit identifies a weakness in supplier controls, the FDA investigator will immediately pivot to Clause 7.4 to verify if the corrective action was effective. The "roadmap" provided by the manufacturer's own internal audit is now the primary tool for the regulator.
Part II: The Legal Architecture of QMSR (21 CFR Part 820)
To fully understand the operational requirements, one must dissect the legal structure of the new regulation. The QMSR uses a method called Incorporation by Reference (IBR), but it is not a "clean" adoption. It is a hybrid regulation that requires simultaneous cross-referencing of the international standard and federal statutes.
2.1 The Supremacy of the FD&C Act
The QMSR incorporates ISO 13485:2016 in its entirety.1 However, 21 CFR 820.1 explicitly states that where a conflict exists between the standard and the Federal Food, Drug, and Cosmetic Act (FD&C Act), the FD&C Act controls.1
This supremacy clause is most critical in the realm of definitions.
- Safety and Performance vs. Safety and Effectiveness: ISO 13485 uses the phrase "safety and performance." The FDA retains the statutory standard of "safety and effectiveness".16 While these concepts overlap, "effectiveness" in the US regulatory context has a specific evidentiary burden (often clinical) that "performance" (which can be engineering-based) may not fully capture.
- Manufacturer: The FDA retains its own definition of "manufacturer" in 21 CFR 820.3. This ensures that entities like specification developers and repackagers/relabelers remain fully within the scope of the regulation, even if they might fall into different categories under a strict reading of ISO definitions.17
2.2 The "Subpart B" Additions (21 CFR 820.35)
The FDA identified specific areas where ISO 13485 did not provide sufficient control to satisfy US statutory mandates. These "gap-fillers" are codified primarily in 21 CFR 820.35 (Control of Records).13 A compliant Quality Manual must explicitly bridge these gaps.
2.2.1 Complaint Handling (The MDR Link)
ISO 13485 Clause 8.2.2 deals with "Complaint Handling," but the requirements are generalized. The FDA has retained prescriptive requirements to ensure alignment with Medical Device Reporting (MDR) under 21 CFR Part 803.16
- Requirement: Manufacturers must record specific data points for every complaint, including the dates of occurrence, the device identification (UDI), and the justification for not investigating a complaint.
- Risk: A system that purely follows ISO 13485 might capture the "nature of the complaint" but miss the specific meta-data required to determine MDR reportability within the strict 30-day (or 5-day) windows.
2.2.2 Servicing Records
The QMSR clarifies the regulatory standing of servicing data. Under 21 CFR 820.35(b), manufacturers must analyze service reports with the same rigor as complaints.16
- Requirement: If a service report represents an event that must be reported under MDR (Part 803), it automatically triggers the complaint handling process.
- Operational Impact: Service technicians often use different software systems (e.g., field service management tools) than the QA department (eQMS). The QMSR demands a validated interface between these systems to ensure that "service" data flows seamlessly into "quality" analysis.
2.2.3 Unique Device Identification (UDI)
The QMSR embeds compliance with 21 CFR Part 830 (UDI) directly into the record-keeping requirements.13
- Requirement: Device History Records (now "Batch Records" under ISO Clause 7.5.1) must capture the UDI of the device. This is not just the lot number; it is the full traceability string.
- Integration: This requires that the ERP or manufacturing execution system (MES) is capable of parsing and storing UDI data in a retrievable format for every batch released.
2.3 Terminology Crosswalk: The Death of the DMR
One of the most visible changes is the retirement of classic FDA nomenclature. The terms Device Master Record (DMR), Design History File (DHF), and Device History Record (DHR) do not appear in the text of the new QMSR.15 They have been replaced by their ISO 13485 equivalents.
Table 1: Terminology Crosswalk (Legacy QSR vs. QMSR)
Analysis: While the terms have changed, the intent remains identical. The FDA has stated that the documentation requirements of the DMR, DHF, and DHR are "adequately described" in the referenced ISO clauses.15 However, manufacturers who hurriedly "Ctrl+F" replaced these terms in their procedures without understanding the subtle scope differences may face findings. FDA investigators will now use ISO terminology during inspections.9 Quality staff must be fluent in translating "Show me your DHF" to "Here is our Design and Development File."
Part III: The New Inspection Paradigm (Compliance Program 7382.850)
The retirement of the Quality System Inspection Technique (QSIT) is arguably the most disruptive change for the "front line" of quality assurance. For over 25 years, QSIT provided a predictable script for inspections. As of February 2, 2026, that script has been shredded.3
It has been replaced by the updated Compliance Program (CP) 7382.850: Inspection of Medical Device Manufacturers.1 This new program is not merely a checklist update; it is a structural reorganization of how the FDA assesses compliance.
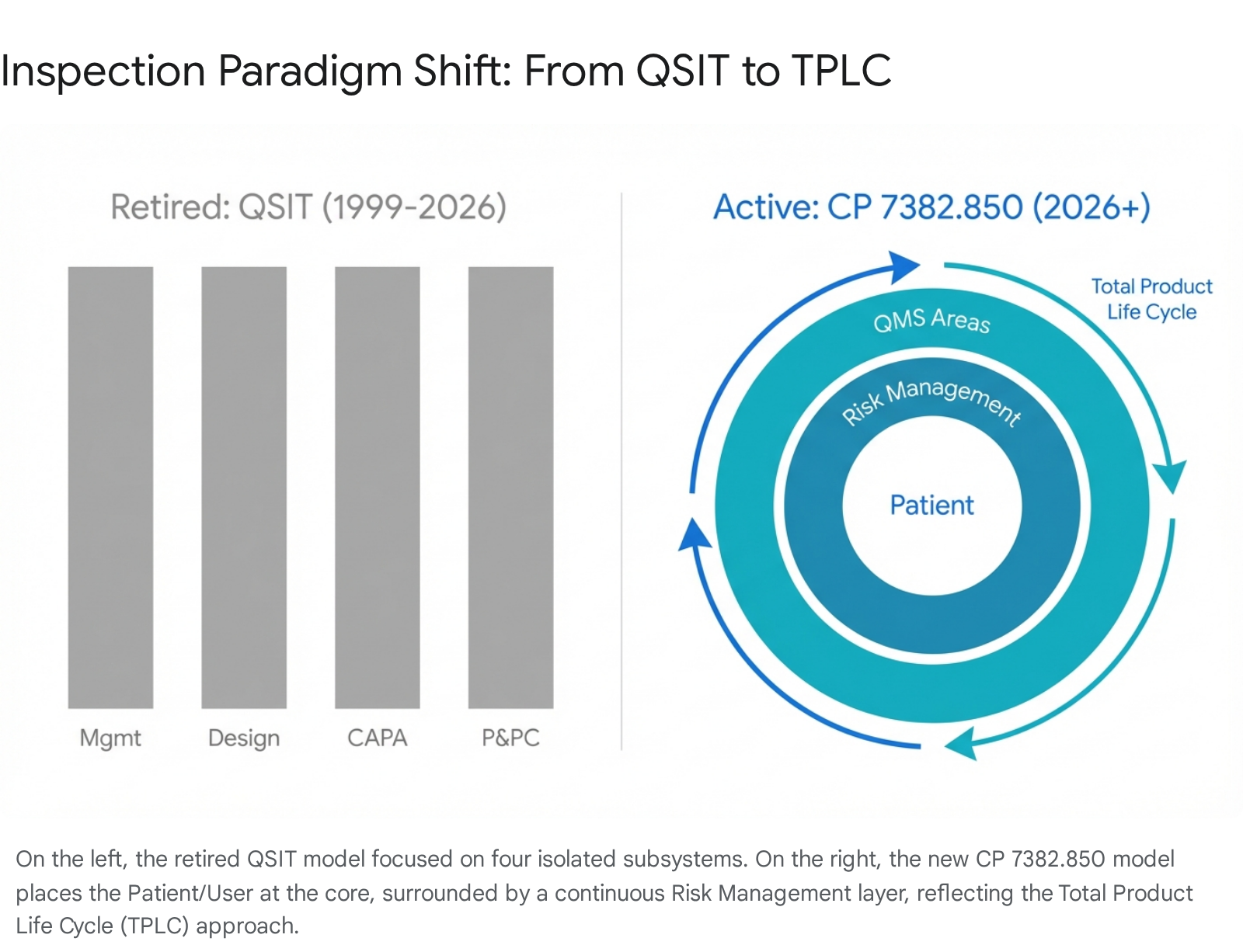
3.1 From Linear Subsystems to TPLC Risk Circles
QSIT used a "top-down" approach focusing on four major subsystems: Management Controls, Design Controls, CAPA, and Production & Process Controls.1 The new CP 7382.850 utilizes a Total Product Life Cycle (TPLC) assessment model.4
The new inspection model organizes requirements into 6 QMS Areas (aligned with ISO 13485 clauses) and 4 Other Applicable FDA Requirements (OAFRs).5
The 6 QMS Areas:
- Management (ISO Clause 5)
- Resources (ISO Clause 6)
- Design and Development (ISO Clause 7.3)
- Production and Service (ISO Clause 7.5)
- Purchasing/Supplier Control (ISO Clause 7.4)
- Measurement, Analysis, and Improvement (ISO Clause 8 - encompassing CAPA)
The 4 OAFRs:
- Medical Device Reporting (MDR) (21 CFR Part 803)
- Corrections and Removals (21 CFR Part 806)
- Registration and Listing (21 CFR Part 807)
- Medical Device Tracking (21 CFR Part 821)
The conceptual shift is best understood visually. The new inspection diagram places "Patients and Users" at the center, surrounded by a "Risk Management" circle.4 This is a profound statement of intent: every inspectional query will be filtered through the lens of risk to the patient. An investigator will not just ask "Did you follow the procedure?" but "Did your deviation from the procedure introduce risk to the patient, and how was that risk assessed?"
3.2 The "Culture of Quality" Assessment
A significant addition to the preamble of the QMSR and the inspection guidance is the FDA's expectation of a "Culture of Quality".5 This is not a "fluff" term; it is an inspectional objective.
Inspectors are trained to look for behaviors and activities that demonstrate top management's commitment. In the old QSIT model, management responsibility was often checked via the "Management Review" dates and attendance. In the new model, investigators will probe deeper:
- Objective Alignment: Are quality objectives linked to patient safety or just business metrics (e.g., "Reduce scrap" vs. "Reduce complaints")?
- Independence: Does the organizational structure support independent quality decision-making, or is Quality overruled by Operations?
- Risk Integration: Is risk management integrated into business decisions, or is it a retrospective paperwork exercise?
The removal of the management review exemption (discussed in Part IV) is the primary tool the FDA will use to assess this culture. If management reviews consistently show "Green" metrics while field complaints are rising, this discrepancy serves as prima facie evidence of a flawed culture of quality.
Part IV: The End of Exemptions (The "Glass House" Era)
Perhaps the most jarring change for legacy Quality Assurance (QA) professionals is the deletion of 21 CFR 820.180(c). This single deletion fundamentally alters the strategy of audit management.
4.1 The Old Rule (Pre-Feb 2, 2026)
Under the QSR, manufacturers were exempt from showing FDA investigators the actual reports of:
- Internal Audits (820.22)
- Management Reviews (820.20(c))
- Supplier Audits (820.50(a))
Manufacturers only had to certify in writing that these activities occurred. This "shield" allowed companies to be brutally honest in their internal findings—listing critical non-conformances without fear that the document itself would become evidence for an FDA observation (Form 483).
4.2 The New Rule (Post-Feb 2, 2026)
The exemption is gone. The QMSR grants FDA investigators full authority to inspect these records.6 The FDA's rationale is that since manufacturers already show these records to ISO auditors (Notified Bodies) and other global regulators (MDSAP), the burden of producing them for the FDA is negligible.6
4.3 Strategic Implications & Risks
This change necessitates a radical shift in how internal documents are authored and managed.
4.3.1 The "Double-Edged Sword" of Internal Audits
Previously, a "good" internal audit was one that found everything. A finding like "Process X is completely broken and poses a high risk" was a badge of honor for a rigorous auditor.
Now, that same statement in a report is a "smoking gun" for an FDA inspector. If an investigator opens an internal audit report from March 2026 and sees a critical finding, their next question will be: "Show me the CAPA."
If the CAPA is open, late, or ineffective, the manufacturer has effectively handed the investigator the evidence for a warning letter. The investigator does not need to dig for the violation; the manufacturer has documented it for them.
- Risk: Companies may be tempted to "water down" internal audit findings to avoid creating liability.
- Counter-Risk: "Watering down" audits leads to a "Culture of Quality" violation. If an FDA investigator finds a problem on the floor that the internal audit should have caught but didn't (or documented vaguely), the finding elevates to a Management Control violation for an ineffective audit program.7
4.3.2 Management Review Minutes
Management reviews often contain sensitive discussions about resource constraints (e.g., "We can't hire more QA staff due to budget cuts"). Under QMSR, these minutes are discoverable.9
- Scenario: A recall occurs due to a missed inspection. The FDA reviews management review minutes and finds a VP of Operations denying a request for more inspectors to save money. This creates a direct line of liability to executive management, supporting enforcement actions under the Park Doctrine (holding executives personally liable for corporate failures).
Actionable Advice:
- Professionalize the Writing: Internal audit reports and management review minutes must now be written with the assumption that they will be read by a prosecutor. Use objective, factual language (e.g., "The sampling plan was not followed in 3 instances") rather than inflammatory language (e.g., "The sampling process is a disaster").
- Close the Loop: Never open an internal audit finding without immediately opening the corresponding correction/corrective action. The gap between "Identification" and "Action" is where enforcement thrives.
Part V: Deep Dive into ISO 13485 Clause Mapping for FDA Compliance
The core of the new QMSR is the incorporation of ISO 13485:2016. However, simply having an ISO-compliant system is insufficient without understanding the specific FDA "overlay." This section provides a granular analysis of how critical ISO clauses map to the new FDA expectations.
5.1 Management Responsibility (ISO Clause 5 vs. Legacy 820.20)
Under the legacy QSR, "Management Responsibility" was a distinct section (820.20) that required a quality policy, organization structure, and management review.
- QMSR Reality: The FDA now enforces ISO Clause 5.
- The "Management Representative" Nuance: ISO 13485 Clause 5.5.2 explicitly requires a "Management Representative." The old QSR 820.20(b)(3) also required this. However, the QMSR preamble emphasizes the "Top Management" involvement (Clause 5.1).5
- Insight: The FDA is moving away from the idea that quality is the job of the "Management Rep" alone. They are adopting the ISO view that Top Management (CEO/President) is accountable.
- Inspection Tactic: In a CP 7382.850 inspection, the investigator will likely interview the CEO/President directly to verify their knowledge of the Quality Policy and Objectives (Clause 5.3, 5.4.1). Ignorance at the top level is now a direct non-conformance against Clause 5.1.
5.2 Resource Management (ISO Clause 6 vs. Legacy 820.20/25)
The legacy QSR had requirements for "Personnel" (820.25). ISO Clause 6 is broader, covering "Provision of Resources," "Human Resources," "Infrastructure," and "Contamination Control."
- The Competency Shift: ISO Clause 6.2 requires determining the necessary competence for personnel performing work affecting product quality. The FDA will scrutinize training records not just for "attendance" (did they sign the sheet?) but for "effectiveness" (did they learn?).9
- Infrastructure & Contamination: ISO Clauses 6.3 and 6.4 provide detailed requirements for maintaining the work environment. For sterile manufacturers, this maps closely to the previous 820.70 controls but adds the specific risk-based environmental control logic of ISO.
5.3 Product Realization (ISO Clause 7 vs. Legacy Design/Production)
This is the largest section, encompassing the old "Design Controls" (820.30), "Purchasing" (820.50), and "Production and Process Controls" (820.70).
5.3.1 Design and Development (Clause 7.3)
The QMSR clarifies that Clause 7.3 applies to the same devices that were subject to 820.30 (Class II, Class III, and select Class I devices).
- Risk in Design: ISO 13485 Clause 7.3.3(c) explicitly requires design outputs to reference acceptance criteria and identify those essential for the safe and proper use of the medical device. This aligns with the FDA's focus on "Essential Outputs."
- The Traceability Matrix: While not explicitly demanded by the text of ISO, the requirement to trace inputs to outputs (7.3.7 verification) remains a standard expectation for FDA inspections to demonstrate validation.
5.3.2 Purchasing (Clause 7.4)
This is a high-risk area. The FDA has historically issued many 483s for "Lack of Supplier Control."
- Risk-Based Control: ISO 13485 Clause 7.4.1 requires that the type and extent of control applied to the supplier be dependent on the risk involved.9
- The Change: Previously, manufacturers might treat all suppliers similarly (e.g., "Annual Survey"). Under QMSR, treating a critical PCB contract manufacturer the same as a cardboard box supplier is a violation of Clause 7.4.1. The "Control" must match the "Risk."
5.4 Measurement, Analysis, and Improvement (ISO Clause 8 vs. Legacy CAPA)
This section replaces the old 820.100 (CAPA), 820.90 (Non-conforming Product), and 820.80 (Acceptance Activities).
5.4.1 Feedback (Clause 8.2.1)
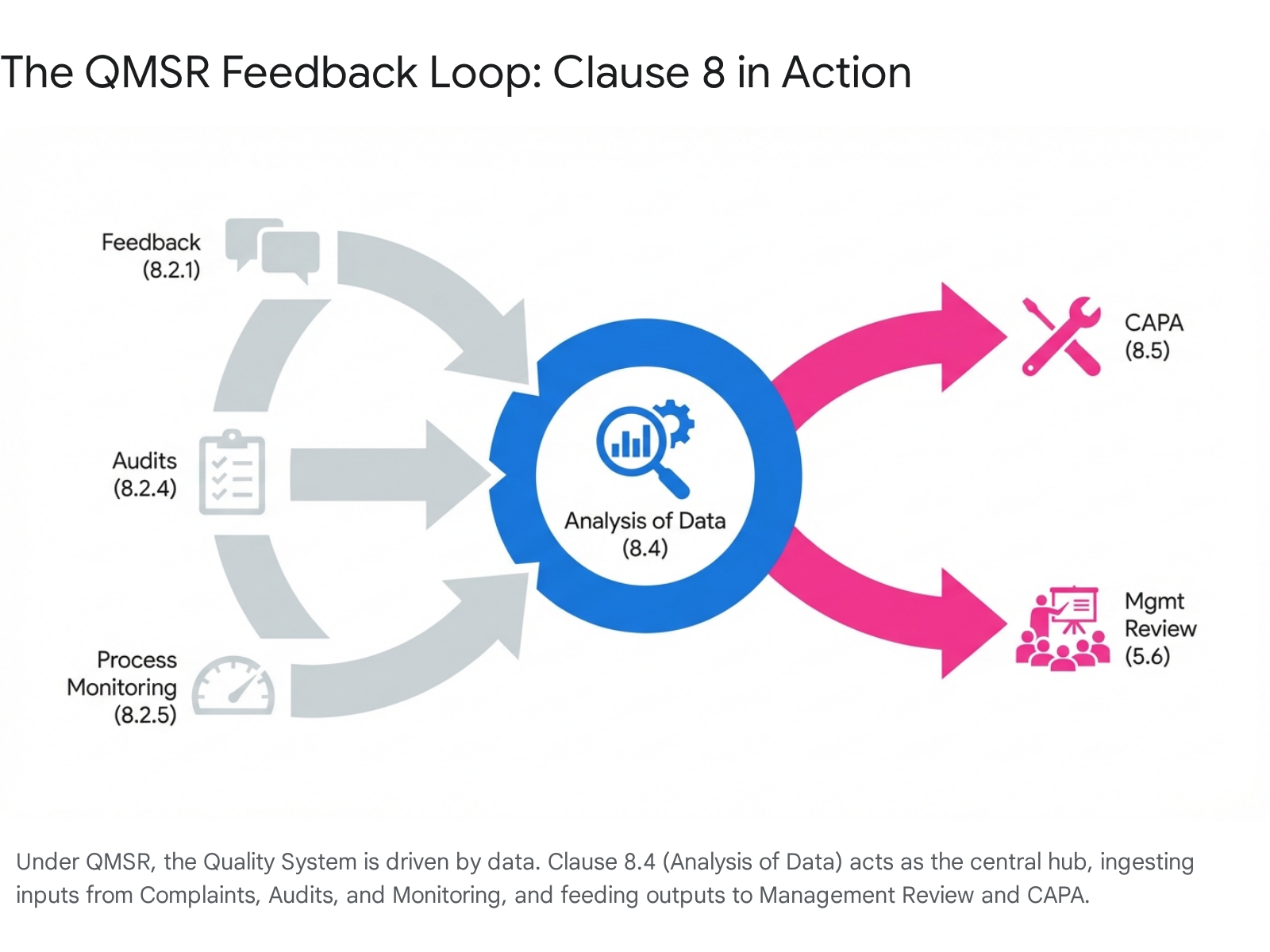
This is a new concept for the FDA regulation. It requires active gathering of information on whether the organization has met customer requirements.24 This goes beyond passive "Complaint Handling." Manufacturers must now show evidence of proactive soliciting of feedback (e.g., surveys, focus groups) as part of their QMS.
5.4.2 Analysis of Data (Clause 8.4)
This is the engine of the QMS. The FDA expects to see statistical techniques (ISO 8.1 / Legacy 820.250) used here to identify trends. The output of Analysis of Data (8.4) is the primary input for Management Review (5.6) and CAPA (8.5). If this linkage is broken—e.g., if data is collected but never analyzed to trigger a CAPA—the system is non-compliant.
5.4.3 Correction, Corrective Action, and Preventive Action
As highlighted in Part I, the separation of these terms is critical.
- Correction (8.2.1): Immediate fix (e.g., rework).
- Corrective Action (8.5.2): Elimination of the cause of a detected non-conformity.
- Preventive Action (8.5.3): Elimination of the cause of a potential non-conformity.
US manufacturers must update their procedures to ensure these are distinct pathways.
Part VI: Global Harmonization Context (FDA & EU)
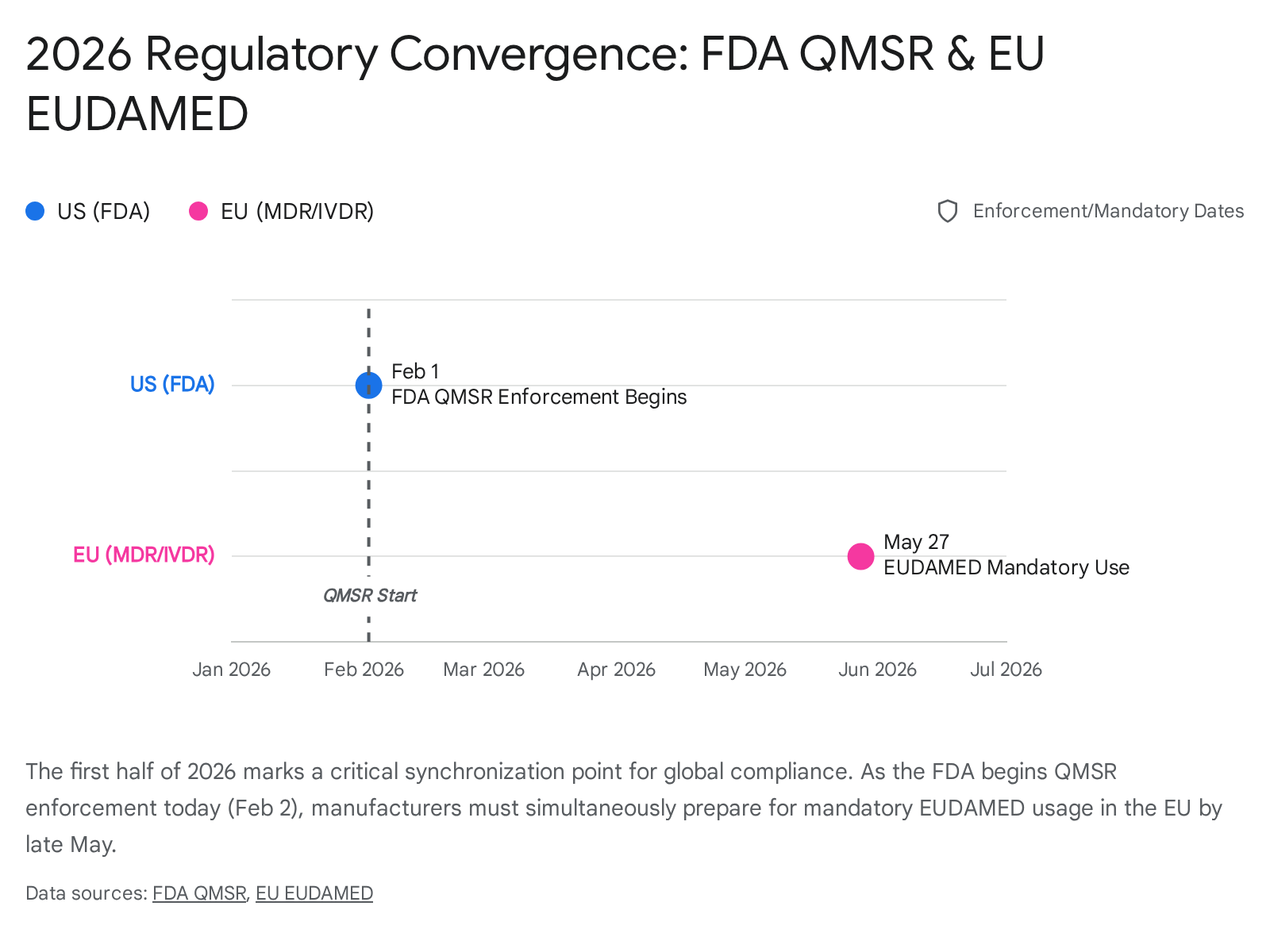
The timing of the QMSR effective date (Feb 2, 2026) is inextricably linked to the global regulatory calendar. The FDA's move to harmonize with ISO 13485 aligns the US with the Medical Device Single Audit Program (MDSAP), used by Canada, Australia, Brazil, and Japan.13 However, the most pressing parallel event is in the European Union.
6.1 The "May 2026" EUDAMED Cliff
While US manufacturers are grappling with QMSR today, they must look ahead to May 28, 2026. On this date, the first four modules of EUDAMED (the European database on medical devices) become mandatory.8
- Mandatory Modules: Actor Registration, UDI/Device Registration, Notified Bodies & Certificates, Market Surveillance.
- Impact: A US manufacturer selling in the EU must have their data ready for upload. The transparency required by EUDAMED (public access to device data) mirrors the transparency now required by FDA inspections (access to internal audits). The "Black Box" of manufacturer data is being pried open on both sides of the Atlantic simultaneously.
6.2 The Transition Crunch
Manufacturers of Class D IVDs (In Vitro Diagnostics) and legacy Medical Device Directive (MDD) devices are also facing transition deadlines in May 2026 and May 2027.26 The convergence of these dates means that Quality/Regulatory (QA/RA) teams are currently stretched to their breaking point.
- Resource Management Risk: FDA auditors inspecting under the "Resources" area (ISO Clause 6) will look for evidence that the QA/RA function is adequately staffed. If a company is failing to meet CAPA timelines because "everyone is working on EUDAMED," this is a valid finding under Clause 6.1 (Provision of Resources).
Part VII: Implementation Guide & Checklist (Feb 2, 2026 Status)
For the qms.coach audience, the following checklist represents the "survival guide" for the immediate post-effective period. This goes beyond the theoretical and addresses the practical steps required this week.
7.1 Immediate Actions (Week 1 of QMSR)
- Stop Using QSIT: Physically remove the QSIT manual from audit preparation rooms. Replace it with CP 7382.850. Ensure that the "Front Room" audit staff are trained to speak to the 6 QMS Areas, not the 4 Subsystems.
- Quarantine Internal Records: Immediately review the "Internal Audit" and "Management Review" folders. Ensure that the records for the current period (starting Feb 2, 2026) are impeccable.
- Note: FDA has stated they can review records created before the effective date if they are part of the current QMS.6 However, the focus will likely be on whether the current system is working.
- Audit the "Bridge": Verify that the procedure mapping ISO 13485 Clause 8.2.1 to 21 CFR 820.35 (Complaint Files) is active and understood by complaint handling staff.
- Verify Terminology in Software: Check the eQMS (Electronic Quality Management System). Do the drop-down menus still say "DHR Review"? Update them to "Batch Record Review" or "Release Review" to match the new SOPs.
7.2 The "Sleep Well" Test
The ultimate test of QMSR readiness is the removal of the fear of discovery.
- Old Mindset: "We hope the FDA doesn't ask for the internal audit report."
- New Mindset: "When the FDA asks for the internal audit report, it will demonstrate that our system detected the issue, assessed the risk, and implemented a corrective action."
Part VIII: Risk Management as a System
The final and most pervasive change is the role of Risk Management. Under the QSR, risk was often treated as a "Design Control" activity (ISO 14971). Under QMSR, risk is the "philosophical centerpiece".9
8.1 Integrated Risk Management
ISO 13485 requires risk management throughout the entire QMS (Clause 4.1.2(b)). This means "Risk" must be a column in:
- Supplier Qualification Matrices: (High risk supplier = On-site audit; Low risk = Questionnaire).
- Training Plans: (High risk task = Effectiveness check; Low risk = Read & Understand).
- Internal Audit Schedules: (High risk process = Audit every 6 months; Low risk = Annual).
If an FDA investigator asks, "Why did you audit this supplier only once in three years?", the answer must be, "Because our risk assessment (Document X) determined they are a low-risk provider of non-critical components." Any other answer (e.g., "We forgot" or "That's our standard policy") is a non-conformance.
Conclusion
The February 2, 2026, effective date of the QMSR is a watershed moment for the US medical device industry. The "harmonization" is now a reality, and the "transition" is over. Manufacturers who are still "preparing" are already late.
The QMSR is not just a new rulebook; it is a new language. It replaces the distinct dialect of the FDA QSR with the global lingua franca of ISO 13485, but with a distinct American accent (Subpart B). Fluency in this language—understanding the nuances of "performance" vs. "effectiveness," "corrective" vs. "preventive," and "compliance" vs. "culture"—is now the baseline requirement for market access in the United States. As of today, the bridge has been crossed. The era of the "Glass House" Quality System has begun.
Works cited
- Quality Management System Regulation (QMSR) - FDA, accessed February 2, 2026, https://www.fda.gov/medical-devices/postmarket-requirements-devices/quality-management-system-regulation-qmsr
- Quality Management System Regulation: Final Rule Amending the Quality System Regulation – Frequently Asked Questions | FDA, accessed February 2, 2026, https://www.fda.gov/medical-devices/quality-system-qs-regulationmedical-device-current-good-manufacturing-practices-cgmp/quality-management-system-regulation-final-rule-amending-quality-system-regulation-frequently-asked
- Overview of Device Regulation | FDA, accessed February 2, 2026, https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/overview-device-regulation
- FDA replaces QSIT with new Compliance Program Manual - Donawa Lifescience, accessed February 2, 2026, https://www.donawa.com/global-news/fda-replaces-qsit-with-new-compliance-program-manual/
- Inspection of Medical Manufacturers - 7382.850 - FDA, accessed February 2, 2026, https://www.fda.gov/media/80195/download
- Quality Management System Regulation – Frequently Asked Questions - FDA, accessed February 2, 2026, https://www.fda.gov/medical-devices/quality-management-system-regulation-qmsr/quality-management-system-regulation-frequently-asked-questions
- 21 CFR 820.180 - Exceptions to the US FDA's Record Requirements, accessed February 2, 2026, https://medicaldeviceacademy.com/21-cfr-820-180-exceptions/
- The EUDAMED four first modules will be mandatory to use as from 28 May 2026, accessed February 2, 2026, https://health.ec.europa.eu/latest-updates/eudamed-four-first-modules-will-be-mandatory-use-28-may-2026-2025-11-27_en
- News - QMS.Coach LLC Coaching Services, accessed February 2, 2026, https://www.qms.coach/tag/news/
- Building Your First QMSR-Compliant QMS from Scratch, accessed February 2, 2026, https://www.qms.coach/building-your-first-qmsr-compliant-qms-from-scratch/
- Preparing for the transition from FDA QSR to QMSR - Rook Quality Systems, accessed February 2, 2026, https://rookqs.com/blog-rqs/preparing-for-the-transition-from-fda-qsr-to-qmsr
- Quality Management System Information for Certain Premarket Submission Reviews Draft Guidance - FDA, accessed February 2, 2026, https://www.fda.gov/media/189345/download
- What's left of the QSR in QMSR? Your guide to the new Part 820 - Greenlight Guru, accessed February 2, 2026, https://www.greenlight.guru/blog/qmsr-your-guide-to-part-820
- QMSR Compliance: Common Mistakes and How to Avoid Them - QMS.Coach, accessed February 2, 2026, https://www.qms.coach/common-qmsr-compliance-mistakes-and-how-to-avoid-them/
- FDA QMSR: QSR, ISO 13485 & Harmonization Explained - Greenlight Guru, accessed February 2, 2026, https://www.greenlight.guru/blog/qmsr-quality-management-system-regulation
- FDA Finalizes Rule Incorporating ISO 13485 into New Quality Management System Regulation (QMSR) | Covington & Burling LLP, accessed February 2, 2026, https://www.cov.com/en/news-and-insights/insights/2024/02/fda-finalizes-rule-incorporating-iso-13485-into-new-quality-management-system-regulation-qmsr
- February 2, 2026 Is Quickly Approaching—Are You QMSR Ready? - Morgan Lewis, accessed February 2, 2026, https://www.morganlewis.com/pubs/2024/10/february-2-2026-is-quickly-approaching-are-you-qmsr-ready
- Remanufacturing and Servicing Medical Devices - FDA, accessed February 2, 2026, https://www.fda.gov/medical-devices/quality-and-compliance-medical-devices/remanufacturing-and-servicing-medical-devices
- Medical Devices; Quality Management System Regulation Technical Amendments, accessed February 2, 2026, https://www.federalregister.gov/documents/2025/12/04/2025-21955/medical-devices-quality-management-system-regulation-technical-amendments
- Top 10 Takeaways from FDA's Revised Quality System Requirements for Medical Devices, accessed February 2, 2026, https://www.ropesgray.com/en/insights/alerts/2024/02/top-10-takeaways-from-fdas-revised-quality-system-requirements-for-medical-devices
- 21 CFR Part 820 -- Quality System Regulation - eCFR, accessed February 2, 2026, https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820
- The new QMSR and the FDA inspections - mdi Consultants, accessed February 2, 2026, https://mdiconsultants.com/the-new-qmsr-and-the-fda-inspections/
- Guide to Inspections of Quality Systems | FDA, accessed February 2, 2026, https://www.fda.gov/files/Guide-to-Inspections-of-Quality-Systems.pdf
- Complete QMSR Compliance Guide - QMS.Coach, accessed February 2, 2026, https://www.qms.coach/complete-qmsr-compliance_guide/
- Overview - Public Health - European Commission, accessed February 2, 2026, https://health.ec.europa.eu/medical-devices-eudamed/overview_en
- Transition periods under the IVDR extended - BioSlice Blog, accessed February 2, 2026, https://www.biosliceblog.com/2024/07/transition-periods-under-the-ivdr-extended/