What to Expect in Your First QMSR Inspection
FDA inspections after February 2, 2026 will look different. Inspectors will reference ISO 13485 clause numbers instead of QSR sections. They'll request records that were previously exempt—management review minutes, internal audit reports, supplier audit documentation.
Reading time: 13 minutes | Last updated: December 2024
FDA inspections after February 2, 2026 will look different. Inspectors will reference ISO 13485 clause numbers instead of QSR sections. They'll request records that were previously exempt—management review minutes, internal audit reports, supplier audit documentation. The regulatory framework they're assessing against has fundamentally changed.
This article prepares you for what to expect when FDA arrives for your first inspection under QMSR.
In this article:
- How QMSR changes FDA inspection approach
- Records you'll need that you didn't before
- Common focus areas and likely questions
- Inspection preparation checklist
- What investigators will be looking for
How QMSR Changes FDA Inspections
The Regulatory Reference Point Changes
Under QSR: Investigators cited 21 CFR 820.xx sections. "We observed a failure to establish and maintain procedures for design input per 820.30(c)."
Under QMSR: Investigators will cite ISO 13485:2016 clauses. "We observed a failure to establish and maintain documented procedures for design input per ISO 13485:2016 Clause 7.3.3."
What this means for you:
- Personnel should know where to find requirements in ISO 13485
- Your procedures should reference ISO 13485 clauses (or map to them)
- You'll need to locate evidence based on ISO clause structure
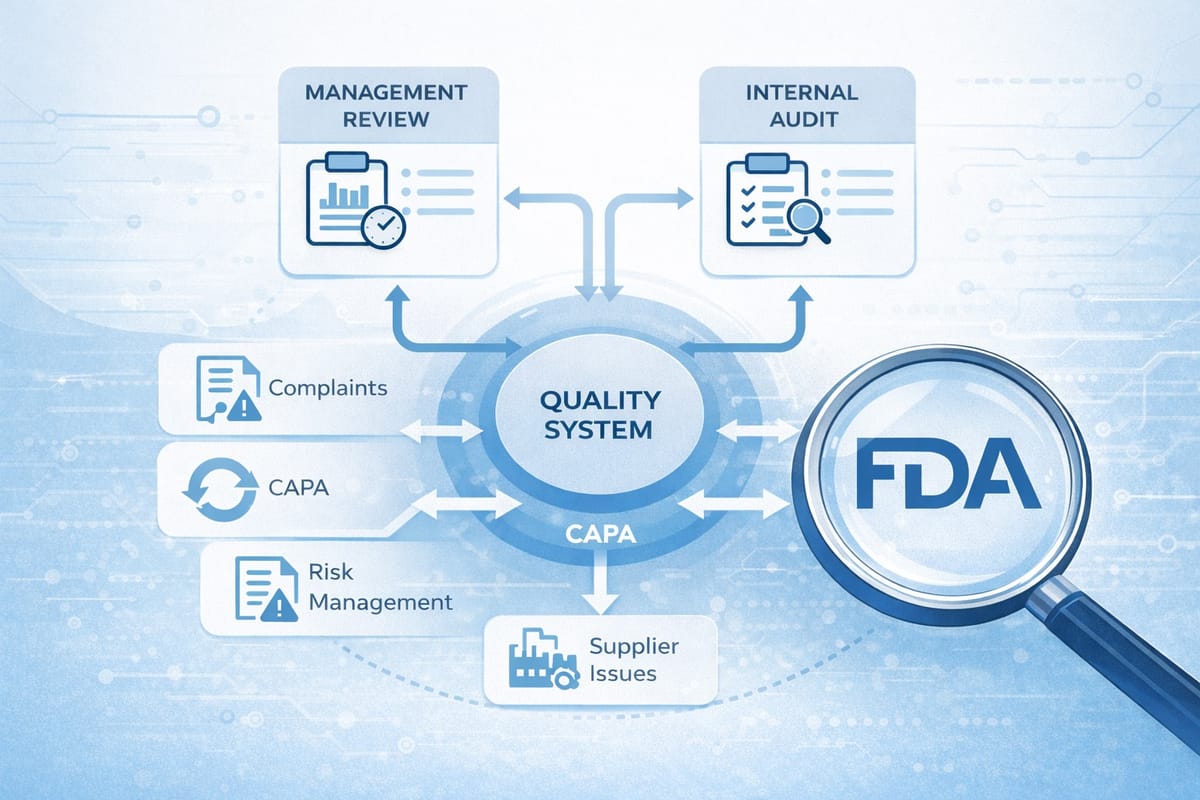
Inspection Scope Expands
Previously exempt records are now fair game:
Management Review Records (Clause 5.6)
- Meeting minutes and attendance
- Input data reviewed
- Decisions and action items
- Follow-up on previous actions
Internal Audit Records (Clause 8.2.4)
- Audit schedule and coverage
- Audit reports and findings
- Corrective action tracking
- Auditor qualification records
Supplier Audit Records (Clause 7.4)
- Supplier audit reports
- Finding documentation
- Corrective action evidence
Risk-Based Thinking Is Expected
Under QSR: Risk management was primarily a design control requirement.
Under QMSR: Investigators may ask about risk-based decisions across your QMS:
- How did you determine this process needed validation?
- Why is this supplier categorized as critical?
- How did you prioritize these CAPAs?
- What risk analysis supported this decision?
Records You'll Need That You Didn't Before
Management Review Package
What to prepare:
- Last 2-3 management review records
- Agenda showing all required inputs
- Minutes documenting discussion and decisions
- Action item log with status and closure evidence
- Supporting data packages (if referenced)
What investigators look for:
- All required inputs addressed (not just listed, but discussed)
- Decisions are documented with rationale
- Actions have owners and due dates
- Follow-up demonstrates systematic closure
Red flags to avoid:
- Boilerplate minutes that don't reflect actual discussion
- Missing inputs or "N/A" without justification
- Action items without documented closure
- Years of identical review content
Internal Audit Documentation
What to prepare:
- Current audit schedule covering all QMS areas
- Audit reports from last 2-3 years
- Finding and observation register
- Corrective action documentation for audit findings
- Auditor training and qualification records
What investigators look for:
- Systematic coverage of all QMS areas over audit cycle
- Clear identification of findings vs. observations
- Timely corrective action initiation and closure
- Evidence of effectiveness verification
- Auditor independence from audited areas
Red flags to avoid:
- Audit schedule not current or not followed
- Findings without documented corrective action
- Long-open corrective actions without justification
- Same findings recurring without escalation
- Auditors assessing their own work
Supplier Audit Records
What to prepare:
- Supplier risk classification rationale
- Audit schedule for suppliers requiring audits
- Audit reports with findings
- Corrective action tracking
- Supplier performance data
What investigators look for:
- Risk-based approach to supplier control
- Audit frequency appropriate to risk
- Findings addressed by suppliers
- Your verification of corrective action effectiveness
Common QMSR Inspection Focus Areas
Based on FDA guidance and ISO 13485 emphasis, expect focus on:
Design Controls (Clause 7.3)
Design control scrutiny continues under QMSR. Expect:
- Design input documentation review
- Traceability to design outputs
- Risk management integration
- Design verification/validation evidence
- Design transfer completeness
Key QMSR difference: Expect questions about risk management integration throughout design, not just at validation.
CAPA (Clauses 8.5.2 and 8.5.3)
Separation verification: Investigators will assess whether you adequately distinguish corrective from preventive action.
Expect questions like:
- "Show me your preventive action procedure."
- "How do you identify potential nonconformities?"
- "What triggers preventive action vs. corrective action?"
- "Show me recent preventive actions and their triggers."
Red flag: If all your "preventive actions" are actually corrections or corrective actions addressing existing problems, expect a finding.
Customer Feedback (Clause 8.2.1)
New area of inquiry: Feedback systems beyond complaint handling.
Expect questions like:
- "How do you collect customer feedback beyond complaints?"
- "Show me your feedback analysis and trending."
- "How does feedback inform your risk management?"
- "How is feedback addressed in management review?"
QMS Software (820.35)
FDA-specific addition: Validation of software used in QMS.
Expect questions like:
- "What software do you use to manage quality activities?"
- "Show me validation documentation for this system."
- "What triggers revalidation?"
- "How do you validate software updates?"
Risk-Based Decisions
Throughout the inspection: Expect questions about risk rationale.
Examples:
- "Why did you decide this process required validation?"
- "How did you determine the appropriate validation extent?"
- "What risk assessment supported this supplier classification?"
- "How do you prioritize CAPA investigations?"
Likely Inspector Questions by Area
Quality Management System (Clause 4)
- "Show me your process interaction documentation."
- "How do you control outsourced processes?"
- "What QMS software do you use and where is the validation?"
- "How do you determine document retention periods?"
Management Responsibility (Clause 5)
- "Who is your top management for QMS purposes?"
- "Show me your quality objectives and how you measure them."
- "May I see your last three management review records?"
- "How are management review actions tracked to closure?"
Resource Management (Clause 6)
- "How do you determine competence requirements?"
- "Show me how you evaluate training effectiveness."
- "How do you ensure work environment requirements are maintained?"
Product Realization (Clause 7)
- "Show me how customer requirements are reviewed before acceptance."
- "What is your customer communication procedure?"
- "How do you determine which suppliers are critical?"
- "What risk assessment supports your validation approach?"
Measurement and Improvement (Clause 8)
- "Describe your customer feedback system."
- "Show me your internal audit schedule and recent reports."
- "What's the difference between your corrective and preventive action procedures?"
- "How do you determine when a CAPA is effective?"
Inspection Preparation Checklist
30 Days Before (Or Whenever You Start)
Documentation preparation:
- [ ] Update Quality Manual to reference QMSR/ISO 13485
- [ ] Verify procedure references are current
- [ ] Organize management review records
- [ ] Organize internal audit records
- [ ] Organize supplier audit records
Gap closure:
- [ ] Complete any open corrective actions
- [ ] Address outstanding audit findings
- [ ] Close or document justification for open management review actions
14 Days Before
Record readiness:
- [ ] Review management review records for completeness
- [ ] Review internal audit records for finding closure
- [ ] Verify CAPA records demonstrate CA/PA separation
- [ ] Confirm QMS software validation documentation exists
Personnel preparation:
- [ ] Brief management on inspection scope changes
- [ ] Train front-line quality on ISO 13485 structure
- [ ] Prepare subject matter experts for interviews
- [ ] Identify document locations and retrieval processes
7 Days Before
Final verification:
- [ ] Conduct mock inspection on high-risk areas
- [ ] Verify all requested documentation can be produced quickly
- [ ] Confirm escort and logistics arrangements
- [ ] Review recent FDA warning letters for common QMSR findings
Day Of Inspection
Execution:
- [ ] Escort investigator professionally
- [ ] Provide requested records promptly
- [ ] Take notes on all requests and questions
- [ ] Clarify questions before responding
- [ ] Don't volunteer information not requested
- [ ] Commit to follow-up for items requiring research
What Investigators Will Be Looking For
Evidence of Systematic Implementation
Not just procedures, but evidence that procedures are followed:
- Records that match procedure requirements
- Consistent execution across similar situations
- Appropriate escalation when problems occur
- Follow-through on commitments
Risk-Based Decision Making
Evidence that risk informs decisions:
- Documented risk rationale for significant decisions
- Proportionate controls based on risk
- Risk consideration in resource allocation
- Periodic risk reassessment
Management Engagement
Evidence that leadership is involved:
- Meaningful management review (not checkbox exercise)
- Resource allocation for quality priorities
- Executive awareness of quality performance
- Accountability for quality outcomes
Continuous Improvement
Evidence that the system improves over time:
- Effective corrective actions (problems don't recur)
- Proactive preventive actions (problems prevented)
- Trending and analysis driving improvement
- Learning from internal and external sources
After the Inspection
If You Receive 483 Observations
- Respond within 15 business days
- Address each observation specifically
- Provide evidence of corrective action taken or planned
- Include timelines for completion
- Don't be defensive; be responsive
If You Receive No Observations
- Don't relax—maintain your systems
- Continue improvement efforts
- Prepare for next inspection cycle
- Document what worked for future reference
Related Resources
- Complete QMSR Compliance Guide 2025-2026
- QMSR vs QSR: 15 Critical Differences You Need to Know
- Common QMSR Compliance Mistakes and How to Avoid Them
- Take the QMSR Readiness Assessment
Want help preparing for your first QMSR inspection?
QMS.Coach provides inspection readiness assessments and mock inspections. Our team has guided organizations through dozens of FDA inspections and can help you identify gaps before investigators do.
Book a Free 15-Minute Consultation →
QMS.Coach LLC | neel@qms.coach