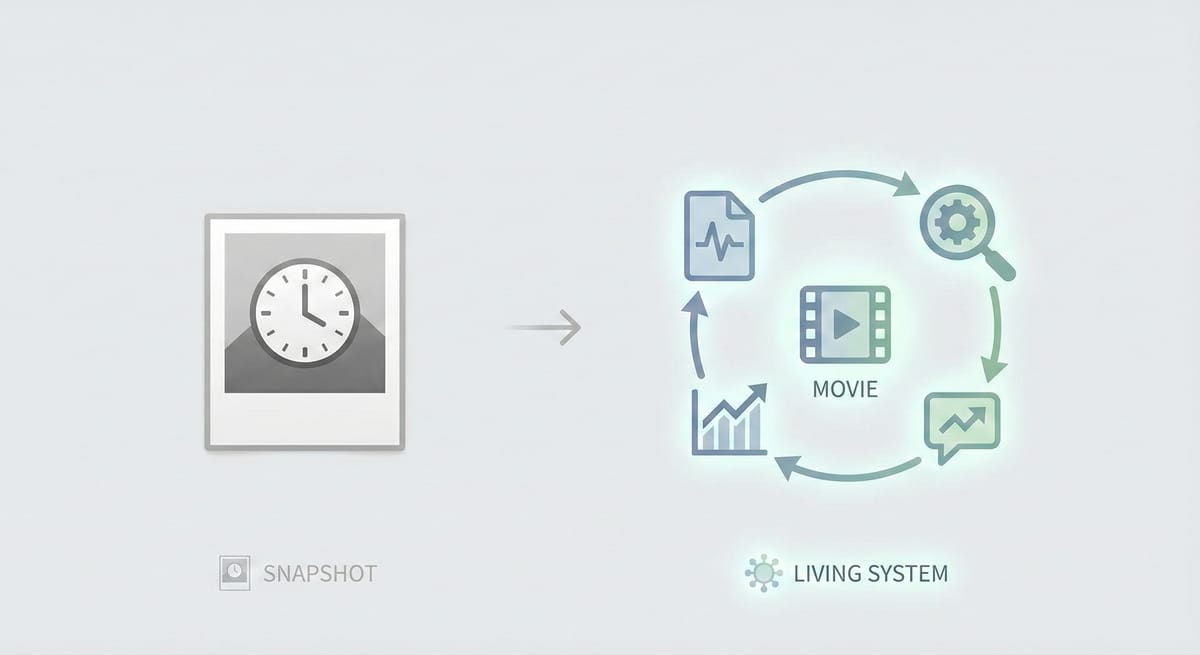
Your FMEA Is a Snapshot. The FDA Wants a Movie.
The companies that are ready for their next inspection aren't the ones with the most sophisticated FMEA software. They're the ones where complaint data flows into the risk file
On February 2, 2026, the QMSR took effect and FDA withdrew the legacy QSIT inspection model.
Last week, one of the first publicly reported post-QMSR FDA inspections wrapped up. Darren Reeves, a medical device quality and regulatory consultant, walked his client through it in real time and shared updates publicly on LinkedIn. His thread — which drew reactions from over 90 quality professionals, including a current FDA investigator — surfaced patterns worth paying attention to.
His takeaway #1:
"Risk, Risk, Risk. Organize your devices by FDA product code by class and highest risk so they don't have to. That is where they will start and you should too."
His takeaway #2:
"HAVE A CURRENT FMEA! Make that document living via your complaints."
That second line stopped me. Not because it's surprising — but because of the word living.
Most FMEAs I've seen aren't living. They're artifacts. Created during design and development, reviewed during internal audits, and largely untouched between those events. The severity scores were assigned three years ago. The occurrence ratings haven't been recalculated since the original design verification. The detection controls reference test methods that have since been updated — or retired.
The FMEA exists. It's just frozen in time.
Under the QMSR's risk-aligned inspection approach, FDA can now probe whether your risk management activities are current and connected to postmarket feedback. An FMEA is one common way to demonstrate that — but not the only acceptable approach. The QMSR does not require ISO 14971 or any specific risk tool; firms may use any appropriately validated risk management process.
What the Investigator Actually Did
Another data point from the same period: a second consultant, F. David Rothkopf, shared that his client's inspection — which started the day after the QMSR deadline — opened with two requests:
- "Let me see your quality plan for QMSR."
- "Let me see your procedure for risk analysis."
From there, the investigator walked through risks related to documentation changes, purchasing evaluation, CAPA risk evaluations, internal audits, and management review. In Rothkopf's words: "He stepped in every QMSR gap to test it out."
What connects both field reports — and these are two datapoints, not a confirmed agency-wide pattern — is that the investigators didn't start with the CAPA log or the complaint files. They started with risk documentation. And they tested whether that documentation reflects current reality, not just whether it exists.
Darren also noted that one investigator reportedly had challenges interpreting ISO 14971, the risk management standard. FDA has publicly stated that staff are being trained on the final rule and the revised inspection process — so this may reflect early-transition growing pains rather than a systemic gap. Still, the fact that risk was the first thing the investigator reached for tells you where the agency's priorities are, even as training continues.
The Snapshot Problem
ISO 14971 requires ongoing risk management activities throughout the product lifecycle. ISO 13485 Clause 7.1 requires that risk management be part of quality planning. Clause 7.3.9 requires that design changes — including those driven by post-production information — be evaluated for their effect on risk.
None of this is new language.
But the enforcement pattern is shifting. Under the old QSR, investigators could — and often did — focus on whether procedures existed and whether records were complete. The FMEA was a document to produce, not a system to interrogate.
Under the QMSR, with ISO 13485 incorporated by reference, a defensible way to prepare is to ensure your risk documentation reflects what's actually happening with your product in the field. In practice, that means:
- Complaint data should be flowing back into your risk analysis
- CAPAs should be updating severity and occurrence ratings when root causes are identified
- Post-market surveillance findings should trigger reassessment of detection controls
- Supplier nonconformances should be evaluated for their impact on the risk profile
The FMEA that was finalized during design transfer and hasn't been touched since? That's a snapshot. And a snapshot of a product that's been on the market for three years doesn't tell the investigator — or your quality team — what the current risk profile actually looks like.
Building the Feedback Loop
The fix isn't rebuilding your FMEA from scratch. It's building a feedback loop so the document updates itself based on the data you're already collecting.
Here's a practical workflow that connects your existing systems:
Step 1: Export your complaint and CAPA data.
Pull 12-24 months of complaint records and closed CAPAs. Most eQMS platforms export to CSV or Excel. You need: failure mode description, product line, root cause (if identified), date, and whether a CAPA was initiated.
Step 2: Categorize failure modes against your existing FMEA.
This is where most teams get stuck — and where AI can help, with important caveats (more on that below).
Map each complaint and CAPA root cause to the failure modes already listed in your FMEA. The question you're answering: Are the things failing in the field the same things we predicted could fail during design?
You'll typically find three buckets:
- Matched: The failure mode is in your FMEA with a severity and occurrence rating. Check whether the real-world data supports those ratings.
- Underscored: The failure mode is in your FMEA, but the occurrence rating is too low based on actual complaint data. Your FMEA says "unlikely" — your complaint data says it happened 14 times last year.
- Missing: The failure mode isn't in your FMEA at all. This is your biggest gap — a real-world risk that your risk analysis doesn't acknowledge.
Step 3: Reassess your risk estimates.
For every failure mode where real-world data contradicts your original assumptions, reassess your risk estimates and controls using whatever method your risk management process defines — whether that's an FMEA with RPNs, a risk matrix, fault tree analysis, or another validated approach. The tool doesn't matter. What matters is that you document the rationale for any changes and that the updated assessment reflects current field data, not original design assumptions.
Step 4: Close the loop in your process.
The one-time exercise above is useful. The sustainable version is a process change:
- Add a step to your CAPA closure procedure: "Has the risk file been reviewed and updated based on this corrective action?" (Yes/No with justification)
- Add a quarterly review trigger: pull new complaint data, compare against FMEA failure modes, flag discrepancies
- Include FMEA currency as a management review input
This is the "living" document Darren was describing. Not a document that someone remembers to update during audit prep — a document that updates as a natural output of your existing quality processes.
Where AI Fits — And Where It Doesn't
The categorization step — mapping hundreds of complaint records to FMEA failure modes — is tedious, time-consuming work. It's also exactly the kind of pattern-matching task that AI handles well.
You can use a general-purpose AI tool (Claude, ChatGPT, Gemini — any of them) to accelerate this step. Feed it your complaint descriptions and your FMEA failure mode list. Ask it to suggest which complaints map to which failure modes, and which complaints don't match anything in your FMEA.
But you need to understand what AI is actually doing here — and what it's not doing.
What AI does well in this context:
- Pattern matching between natural-language complaint descriptions and technical failure mode descriptions
- Identifying clusters — "these 23 complaints all describe variations of the same failure mode"
- Flagging gaps — "these 8 complaints don't map to any existing failure mode in your FMEA"
- Processing volume — categorizing 500 complaints in minutes instead of weeks
What AI does NOT do — and what you must not delegate:
- Risk scoring. AI should never assign severity, occurrence, or detection ratings. Those are engineering judgments that require domain expertise, clinical context, and regulatory understanding. An AI doesn't know the difference between a cosmetic defect and a patient safety risk in your specific device context.
- Root cause determination. AI can suggest categories. It cannot determine why something failed. Root cause analysis requires investigation, not inference.
- Regulatory interpretation. AI can reference ISO 14971 or ISO 13485 clause text. It cannot tell you what an FDA investigator will accept as adequate risk management for your specific product. That's a judgment call that requires regulatory experience.
The hallucination risk is real. AI models generate plausible-sounding text. They can confidently map a complaint to the wrong failure mode, invent a regulatory citation that doesn't exist, or suggest a risk rating that sounds reasonable but has no engineering basis. Every AI-generated mapping must be reviewed by someone who understands the product, the process, and the regulatory requirements.
The privacy and IP risk is real too. Your complaint data contains product-specific failure information, customer details, and potentially regulated information (MDR-reportable events, for example). Before uploading complaint data to any AI platform, understand:
- Where the data is processed and stored
- Whether the platform uses your data for model training
- Whether your use complies with your own data governance policies and any applicable regulatory requirements
- Whether PHI, PII, or trade secrets are present in the data you're uploading
For companies with sensitive complaint data, consider using an on-premise or private-instance AI deployment, or anonymize the data before processing. Remove patient identifiers, customer names, serial numbers, and any MDR reference numbers before the data touches an external AI system.
The governance question: If you use AI to assist with risk categorization, document it. Your quality system should be able to answer: What tool was used? What was its role in the analysis? Who reviewed and approved the output? What was the verification method?
This isn't bureaucracy — it's audit defensibility. When an investigator asks how you updated your risk file, "AI did it" is not an acceptable answer. "We used an AI tool to categorize complaint patterns, then a cross-functional team reviewed the output, validated the mappings against engineering specifications, and made risk scoring decisions based on clinical and regulatory context" — that's an acceptable answer.
A Practical Prompt Template
If you decide to use AI for the categorization step, here's a starting point. This is a template — adapt it to your product and your FMEA structure.
I have a medical device FMEA with the following failure modes: [paste your failure mode list]. I also have complaint records from the past 12 months: [paste complaint descriptions — anonymized, no patient data, no serial numbers]. For each complaint, suggest which FMEA failure mode it most closely relates to. If a complaint doesn't match any existing failure mode, flag it as "UNMATCHED — potential new failure mode." Do not assign risk scores. Do not suggest severity or occurrence ratings. Only categorize.
The key constraints in that prompt:
- "Do not assign risk scores" — keeps the AI in its lane
- "Only categorize" — prevents the model from overstepping into engineering judgment
- Anonymized data — no patient information, no identifying details
After the AI returns its categorization, your team reviews every mapping. Reject the ones that don't make sense. Investigate the unmatched complaints — those are your FMEA gaps.
What "Good" Looks Like Before Your Next Inspection
Based on what we're hearing from the field in these first post-QMSR inspections:
- Your devices are organized by FDA product code and risk class. The investigator will start with your highest-risk products. Have that hierarchy ready — don't make them build it from your DHFs.
- Your FMEA for each high-risk product reflects current field data. Not the original design assumptions — the actual complaint and CAPA history mapped back to failure modes.
- You can trace the path from a complaint through investigation through risk file update. If a CAPA closed last quarter and the risk file wasn't reviewed, that's a gap an investigator will find.
- Your risk management process describes how you make decisions. Darren noted a challenge around ISO 14971 interpretation during the inspection. Know your risk acceptance criteria. Know how you justify residual risk. Be ready to walk an investigator through a decision — not just show them the matrix.
- If you used AI in any part of this process, you can explain exactly how. The tool, the scope of its use, the human review that followed, and the documented decision trail.
The Bottom Line
The QMSR didn't change what good risk management looks like. It changed what gets enforced.
The companies that are ready for their next inspection aren't the ones with the most sophisticated FMEA software. They're the ones where complaint data flows into the risk file, where CAPAs update risk assessments, and where the FMEA reflects what's actually happening — not what was predicted three years ago.
That's the difference between a snapshot and a movie.
And when the investigator walks in and says "let me see your procedure for risk analysis" — the answer isn't a binder. It's a system that moves.
Neel Tiwari is the founder of QMS.Coach, where he helps medical device companies build quality systems that hold up under scrutiny. For questions about QMSR readiness or risk management methodology, reach out at neel@qms.coach.